BioUnfold #14 — Similarity Is Not Mechanism: Limits of Representation in Biology
Self-supervised learning has become the default strategy for training foundation models in biology. It offers structure without labels, scale without annotation, and a way to align heterogeneous assays within a single representation. Yet self-supervision is not a single idea. It is a collection of architectural decisions that encode assumptions about how variation should be organized.
Most discussions focus on data scale or modality. Fewer acknowledge that the architecture itself determines which biological signals are preserved, which are suppressed, and which appear more meaningful than they are. Biological images carry multiple sources of variation at once — morphology, texture, dose response, artifacts, and context — so architectural choices inevitably shape what the model represents.
Architecture is not neutral. It makes a bet about the biology.
The Two Levers of Self-Supervision
Self-supervised models vary along two fundamental axes.
1. The task
This defines what the model predicts without labels.
- Reconstruction tasks (MAE, iBOT) fill in masked pixels or tokens.
- View-alignment tasks (DINO) align representations of augmented views.
- Self-distillation tasks (DINO, MiniLM-like methods) train a student to match a teacher’s embedding space.
2. The regularization
This governs the geometry of the latent space.
- Invariances to augmentations
- Uniformity over a manifold
- Geometric or structural constraints
- Smoothness or sparsity penalties
These distinctions also appear in natural language. Masked language modeling (BERT, RoBERTa) resembles reconstruction, emphasizing local structure. Contrastive and distillation-based approaches (SimCSE, Sentence-BERT, MiniLM) resemble alignment tasks, emphasizing global invariances. Across fields, the task largely determines the structure of the representation.
MAE and DINO Emphasize Different Biology
This divergence is especially visible in imaging.
Reconstruction-based models (MAE, iBOT):
- Capture local structure such as texture, membranes, and organelles.
- Perform well with moderate-scale datasets.
- Learn the physical structure of images before higher-level variation.
Alignment and distillation models (DINO):
- Capture global morphology and invariant features.
- Benefit from large-scale datasets.
- Often amplify structured artifacts when trained on Cell Painting data (illumination gradients, staining drift, debris).
Increasing model size does not guarantee better biological generalization. Often, the model generalizes the most visually dominant structure rather than the underlying biology.
How Embeddings Are Used in Practice
A common pattern has emerged across industry:
- Start with a general-purpose vision backbone such as DINOv2.
- Apply it to biological images without fine-tuning.
- Evaluate similarity or clustering on top of the embeddings.
This frequently works because these evaluations reward visual coherence, not mechanism. The results often look intuitive and therefore seem reliable, but unsupervised success mostly reflects architectural priors rather than biological equivalence.
Why Latent Space Arithmetic Breaks in Biology
Some NLP and vision models exhibit vector arithmetic in embedding space. Biology rarely does. Perturbations follow nonlinear dose–response curves, trigger stress pathways at high concentrations, or produce weak signal at low concentrations. Cell-line and environmental contexts reshape these relationships further.
To analyze this rigorously, let:
- $P_a$ and $P_b$ be two perturbations
- $c$ be the measurement context (cell line, assay condition)
- $f(P \mid c)$ be the embedding function
This notation clarifies how similarity is quantified.
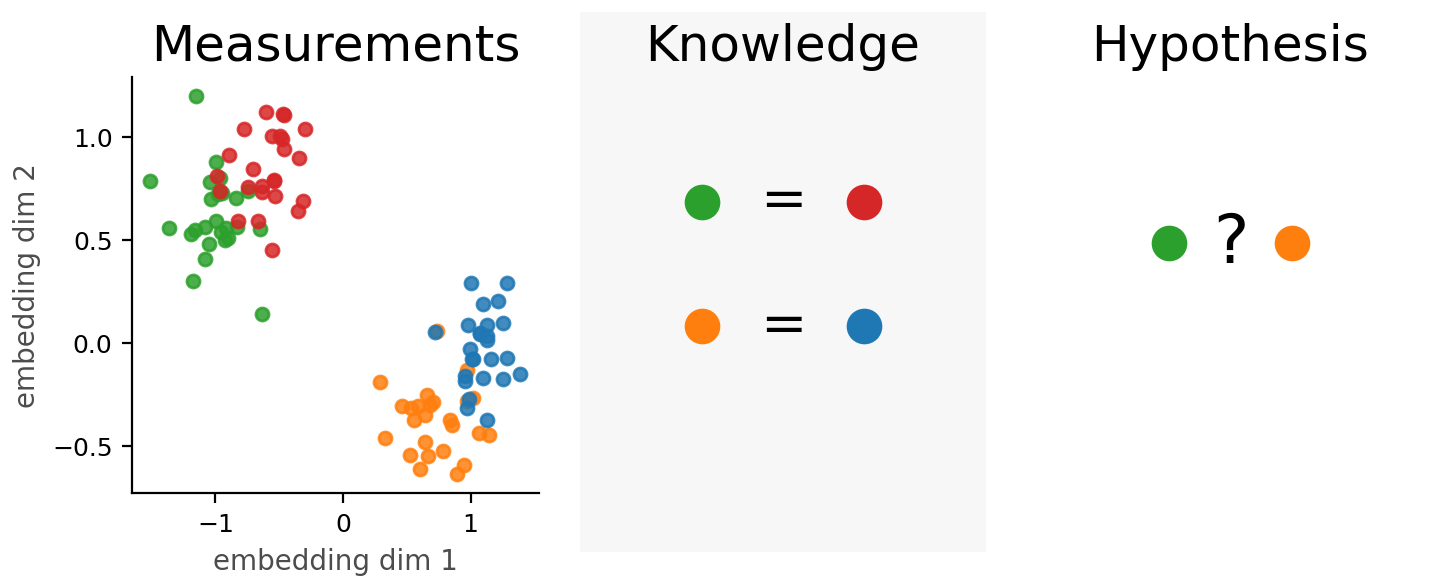
What Biological Workflows Actually Require: Equivalence
The central question in many discovery workflows is:
Do two perturbations produce the same mechanism of action in this context?
Similarity is often assessed using cosine similarity, which measures the angle between embedding vectors rather than their distance. It evaluates whether two perturbations point in the same direction in representation space.
\[\cos(f(P_a \mid c), f(P_b \mid c)) \approx 1 \quad \Rightarrow \quad \text{similar mechanism}\]Cosine similarity assumes that:
- phenotypes scale linearly with dose
- higher concentrations move the embedding in a consistent direction
- the dominant axis of variation corresponds to mechanism
These assumptions often fail. Dose–response curves are nonlinear or biphasic. High doses introduce stress phenotypes unrelated to mechanism. Low doses may show little signal despite true mechanistic similarity. Context shifts alter phenotypes in ways cosine geometry cannot express.
As a result, two perturbations may appear similar at one dose and dissimilar at another. The issue is not model failure but the mismatch between cosine geometry and biological reality.
A more realistic framing introduces a learned equivalence function:
\[g(P_a, P_b \mid c) \approx 0\]This function evaluates whether two perturbations are functionally similar within the same context, without relying on linearity in embedding space.
Embeddings represent perturbations. They do not inherently represent relationships between perturbations. Equivalence must be learned.
The Core Argument
Self-supervised architectures embed assumptions about how biological variation should be organized. They determine:
- which variations cluster
- when similarity reflects appearance rather than mechanism
- how artifacts become embedded as structure
- whether mechanistic equivalence is representable at all
Biological foundation models will not succeed through scale alone. They will succeed when architectural choices align with the biological questions that matter most:
Are two perturbations equivalent in this context?